第7章 BLAST¶
嗨,每个人都喜欢BLAST,对吧?我是指,通过BLAST把你的序列和世界上已知的序列 比较是多么简单方便啊。不过,这章当然不是讲Blast有多么酷,因为我们都已经 知道了。这章是来解决使用Blast的一些麻烦地方——处理大量的BLAST比对结果数据 通常是困难的,还有怎么自动运行BLAST序列比对。
幸运的是,Biopython社区的人早就了解了这些难处。所以,他们已经发展了很多 工具来简化BlAST使用和结果处理。这章会具体讲解怎么用这些工具来做些有用的 事情。
使用BLAST通常可以分成2个步。这两步都可以用上Biopython。第一步,提交你的查询 序列,运行BLAST,并得到输出数据。第二步,用Python解析BLAST的输出,并作进一步 分析。
你第一次接触并运行BLAST也许就是通过NCBI的web服务。事实上,你可以通过多种方式 (这些方式可以分成几类)来使用BLAST。这些方式最重要的区别在于你是在你的自己 电脑上运行一个本地BLAST,还是在远程服务器(另外一台电脑,通常是NCBI的服务器)上运行。 我们将在一个Python脚本里调用NCBI在线BLAST服务来开始这章的内容。
注意: 接下来的 第 8 章介绍的 Bio.SearchIO 是一个
Biopython实验性质的模块。我们准备最终用它来替换原来的 Bio.Blast 模块。
因为它提供了一个更为通用的序列搜索相关的框架。不过,在这个模块的稳定版本发布之前,在实际工作中的代码里,请继续用 Bio.Blast
模块来处理NCBI BLAST。
7.1 通过Internet运行BLAST¶
我们使用 Bio.Blast.NCBIWWW 模块的函数 qblast() 来调用在线版本的BLAST。
这个函数有3个必需的参数:
- 第一个参数是用来搜索的blast程序,这是小写的字符串。对这个参数的选项和描述可以在
http://www.ncbi.nlm.nih.gov/BLAST/blast_program.shtml.
查到。目前
qblast只支持 blastn, blastp, blastx, tblast 和 tblastx. - 第二个参数指定了将要搜索的数据库。同样地,这个参数的选项也可以在 http://www.ncbi.nlm.nih.gov/BLAST/blast_databases.shtml. 查到
- 第三个参数是一个包含你要查询序列的字符串。这个字符串可以是序列的本身 (fasta格式的),或者是一个类似GI的id。
qblast 函数还可以接受许多其他选项和参数。这些参数基本上类似于你在BLAST网站页面
能够设置的参数。在这里我们只强调其中的一些:
qblast函数可以返回多种格式的BLAST结果。你可以通过可选参数format_type指定格式关键字为:"HTML","Text","ASN.1", 或"XML"。默认 格式是"XML",这是解析器期望的格式, 7.3 节对其有详细的描述。- 参数
expect指定期望值,即阀值 e-value。
更多可选的BLAST参数,请参照NCBI的文档,或者是Biopython内置的文档。
>>> from Bio.Blast import NCBIWWW
>>> help(NCBIWWW.qblast)
...
请注意,NCBI BLAST 网站上的默认参数和QBLAST的默认参数不完全相同。如果你得到了 不同的结果,你就需要检查下参数设置 (比如,e-value阈值和gap值).
举个例子,如果你有条核酸序列,想使用BLAST对核酸数据库(nt)进行搜索,已知这条查询序列的GI号, 你可以这样做:
>>> from Bio.Blast import NCBIWWW
>>> result_handle = NCBIWWW.qblast("blastn", "nt", "8332116")
或者,我们想要查询的序列在FASTA文件中,那么我们只需打开这个文件并把这条记录读入到字符串,然后用这个字符串作为查询参数:
>>> from Bio.Blast import NCBIWWW
>>> fasta_string = open("m_cold.fasta").read()
>>> result_handle = NCBIWWW.qblast("blastn", "nt", fasta_string)
我们同样可以读取FASTA文件为一个 SeqRecord 序列对象,然后以这个序列自身作为参数:
>>> from Bio.Blast import NCBIWWW
>>> from Bio import SeqIO
>>> record = SeqIO.read("m_cold.fasta", format="fasta")
>>> result_handle = NCBIWWW.qblast("blastn", "nt", record.seq)
只提供序列意味着BLAST会自动分配给你一个ID。你可能更喜欢用 SeqRecord
对象的format方法来包装一个fasta字符串,因为这个对象会包含fasta文件中已有的ID
>>> from Bio.Blast import NCBIWWW
>>> from Bio import SeqIO
>>> record = SeqIO.read("m_cold.fasta", format="fasta")
>>> result_handle = NCBIWWW.qblast("blastn", "nt", record.format("fasta"))
如果你的序列在一个非FASTA格式的文件中并且你用 Bio.SeqIO (看第 5 章)
把序列取出来了,那么这个方法更有用。
不论你给 qblast() 函数提供了什么参数,都应该返回一个handle object的结果(
默认是XML格式)。下一步就是将这个XML输出解析为代表BLAST搜索结果的Python
对象( 7.3 )。
不过,也许你想先把这个XML输出保存一个本地文件副本。当调试从BLAST结果提取信息的代码的时候,我发现这样做
尤其有用。(因为重新运行在线BLAST搜索很慢并且会浪费NCBI服务器的运行时间)。
这里我们需要注意下:因为用 result_handle.read() 来读取BLAST结果只能用一次 -
再次调用 result_handle.read() 会返回一个空的字符串.
>>> save_file = open("my_blast.xml", "w")
>>> save_file.write(result_handle.read())
>>> save_file.close()
>>> result_handle.close()
这些做好后,结果已经存储在 my_blast.xml 文件中了并且原先的handle中的数据
已经被全部提取出来了(所以我们把它关闭了)。但是,BLAST解析器的 parse 函数(描述见 7.3 )
采用一个文件句柄类的对象,所以我们只需打开已经保存的文件作为输入。
>>> result_handle = open("my_blast.xml")
既然现在已经把BLAST的结果又一次读回handle,我们可以分析下这些结果。所以我们正好可以去读 关于结果解析的章节(看下面 7.3 )。你现在也许想跳过去看吧 …
7.2 本地运行BLAST¶
7.2.1 介绍¶
在本地运行BLAST(跟通过internet运行比,见 7.1 ) 至少有2个主要优点:
- 本地运行BLAST可能比通过internet运行更快;
- 本地运行可以让你建立自己的数据库来对序列进行搜索。
处理有版权的或者没有发表的序列数据也许是本地运行BLAST的另一个原因。你也许 不能泄露这些序列数据,所以没法提交给NCBI来BLAST。
不幸的是,本地运行也有些缺点 - 安装所有的东东并成功运行需要花些力气:
- 本地运行BLAST需要你安装相关命令行工具。
- 本地运行BLAST需要安装一个很大的BLAST的数据库(并且需要保持数据更新).
更令人困惑的是,至少有4种不同的BLAST安装程序包,并且还有其他的一些工具能 产生类似的BLAST 输出文件,比如BLAT。
7.2.2 单机版的NCBI老版本BLAST¶
NCBI “老版本” BLAST
包括命令行工具 blastall , blastpgp 和 rpsblast 。
这是NCBI发布它的替代品BLAST+ 前使用最为广泛的单机版BLAST工具。
Bio.Blast.Applications 模块有个对老版本NCBI BLAST 工具像 blastall , blastpgp
和 rpsblast 的封装, 并且在 Bio.Blast.NCBIStandalone 还有个辅助函数。
这些东东现在都被认为是过时的,并且当用户们迁移到BLAST+程序套件后,这些都会被弃用,
最终从Biopython删除。
为了减少你的困惑,我们在这个指南中不会提到怎么从Biopython调用这些老版本的工具。
如果你有兴趣,可以看下在Biopython 1.52中包含的基本指南。(看下 biopython-1.52.tar.gz
或者 biopython-1.52.zip 中Doc目录下的指南的PDF文件 或者 HTML 文件)。
7.2.3 单机版 NCBI BLAST+¶
NCBI “新版本”的
BLAST+
在2009年发布。它替代了原来老版本的BLAST程序包。Bio.Blast.Applications 模块
包装了这些新工具像 blastn , blastp , blastx , tblastn , tblastx
(这些以前都是由 blastall 处理)。而 rpsblast 和 rpstblastn (替代了
原来的 rpsblast )。我们这里不包括对 makeblastdb 的包装,它在BLAST+中用于从FASTA文件
建立一个本地BLAST数据库,还有其在老版本BLAST中的等效工具 formatdb 。
这节将简要地介绍怎样在Python中使用这些工具。如果你已经阅读了并试过 6.4 节的序列联配(alignment)工具,下面介绍 的方法应该是很简单直接的。首先,我们构建一个命令行字符串(就像你使用单机版 BLAST的时候,在终端打入命令行一样)。然后,我们在Python中运行这个命令。
举个例子,你有个FASTA格式的核酸序列文件,你想用它通过BLASTX(翻译)来搜索 非冗余(NR)蛋白质数据库。如果你(或者你的系统管理员)下载并安装好了这个数据库, 那么你只要运行:
blastx -query opuntia.fasta -db nr -out opuntia.xml -evalue 0.001 -outfmt 5
这样就完成了运行BLASTX查找非冗余蛋白质数据库,用0.001的e值并产生XML格式的 输出结果文件(这样我们可以继续下一步解析)。在我的电脑上运行这条命令花了大约6分钟 - 这就是为什么我们需要保存输出到文件。这样我们就可以在需要时重复任何基于这个输出的分析。
在Biopython中,我们可以用NCBI BLASTX包装模块 Bio.Blast.Applications 来构建
命令行字符串并运行它:
>>> from Bio.Blast.Applications import NcbiblastxCommandline
>>> help(NcbiblastxCommandline)
...
>>> blastx_cline = NcbiblastxCommandline(query="opuntia.fasta", db="nr", evalue=0.001,
... outfmt=5, out="opuntia.xml")
>>> blastx_cline
NcbiblastxCommandline(cmd='blastx', out='opuntia.xml', outfmt=5, query='opuntia.fasta',
db='nr', evalue=0.001)
>>> print blastx_cline
blastx -out opuntia.xml -outfmt 5 -query opuntia.fasta -db nr -evalue 0.001
>>> stdout, stderr = blastx_cline()
在这个例子中,终端里应该没有任何从BLASTX的输出,所以stdout和stderr是空的。
你可能想要检查下输出文件 opuntia.xml 是否已经创建。
如果你回想下这个指南的中的早先的例子,opuntia.fasta 包含7条序列,
所以BLAST XML 格式的结果输出文件应该包括多个结果。因此,我们在
下面的 7.3 节将用 Bio.Blast.NCBIXML.parse() 来
解析这个结果文件。
7.2.4 WU-BLAST 和 AB-BLAST¶
你也许会碰到 Washington University BLAST (WU-BLAST),
和它的后继版本`Advanced Biocomputing BLAST <http://blast.advbiocomp.com>`__ (AB-BLAST,
在2009年发布,免费但是没有开源)。这些程序包包括了命令工具行
wu-blastall 和 ab-blastall 。
Biopython 目前还没有提供调用这些工具的包装程序,但是应该可以解析它们 与NCBI兼容的输出结果。
7.3 解析BLAST 输出¶
就像上面提过的那样,BLAST能生成多种格式的输出,比如 XML, HTML 和纯文本格式。 以前,Biopython有针对HTML 和纯文本格式输出文件的解析器,因为当时只有这两种 格式的输出结果文件。不幸的是,这两种方式的BLAST 输出结果一直在变动,而每次 变动就会导致解析器失效。 所以,我们删除了针对HTML格式的解析器,不过纯文本格式 的解析还可以用(见 7.5 )。使用这个解析器 有一定的风险,它可能能工作也可能无效,依赖于你正在使用哪个BLAST版本。
跟上BLAST输出文件格式的改变很难,特别是当用户使用不同版本的BLAST的时候。 我们推荐使用XML格式的解析器。因为最近版本的BLAST能生成这种格式的输出结果。 XML格式的输出不仅比HTML 和纯文本格式的更稳定,而且解析起来更加容易自动化, 从而提高整个Biopython整体的稳定性。
你可以通过好几个途径来获得XML格式的BLAST输出文件。对解析器来说,不管你是 怎么生成输出的,只要是输出的格式是XML就行。
- 你可以通过Biopython来运行因特网上的BLAST,就像 7.1 节描述的那样。
- 你可以通过Biopython来运行本地的BLAST,就像 7.2 节描述的那样。
- 你可以在通过浏览器在NCBI网站上进行BLAST搜索,然后保存结果文件。你需要选择输出 结果文件是XML格式的,并保存最终的结果网页(你知道,就是包含所有有趣结果的那个网页) 到文件。
- 你也可以直接运行本地电脑上的BlAST,不通过Biopython,保存输出结果到文件。 同样的你也需要选择输出文件格式为XML。
关键点就是你不必用Biopython脚本来获取数据才能解析它。通过以上任何一种方式
获取了结果输出,你然后需要获得文件句柄来处理它。在Python中,一个文件句柄就是一种
用于描述到任何信息源的输入的良好通用的方式,以便于这些信息能够使用 read() 和 readline()
函数(见章节 22.1 )来获取。
如果你一直跟着上几节用来和BLAST交互的代码的话,你已经有了个 result_handle
,一个用来得到BLAST的结果文件句柄。 比如通过GI号来进行一个在线BLAST搜索:
>>> from Bio.Blast import NCBIWWW
>>> result_handle = NCBIWWW.qblast("blastn", "nt", "8332116")
如果你通过其他方式运行了BLAST,并且XML格式的BLAST结果输出文件是 my_blast.xml ,
那么你只需要打开文件来读:
>>> result_handle = open("my_blast.xml")
好的,现在我们已经有了个文件句柄,可以解析输出结果了。解析结果的代码 很短。如果你想要一条BLAST输出结果(就是说,你只用了一条序列去搜索):
>>> from Bio.Blast import NCBIXML
>>> blast_record = NCBIXML.read(result_handle)
或者, 你有许多搜索结果(就是说,你用了多条序列去BLAST搜索)
>>> from Bio.Blast import NCBIXML
>>> blast_records = NCBIXML.parse(result_handle)
就像 Bio.SeqIO 和 Bio.AlignIO (参见第 5
和第 6 章), 我们有一对输入函数, read 和
parse 。 当你只有一个输出结果的时候用 read 。当你有许多
输出结果的时候,可以用 parse 这个迭代器。 但是,我们调用函数获得结果
不是 SeqRecord 或者 MultipleSeqAlignment 对象,我们得到BLAST记录对象。
为了能处理BLAST结果文件很大有很多结果这种情况, NCBIXML.parse()
返回一个迭代器。简单来说,一个迭代器可以让你一个接着一个地获得BLAST
的搜索结果。
>>> from Bio.Blast import NCBIXML
>>> blast_records = NCBIXML.parse(result_handle)
>>> blast_record = blast_records.next()
# ... do something with blast_record
>>> blast_record = blast_records.next()
# ... do something with blast_record
>>> blast_record = blast_records.next()
# ... do something with blast_record
>>> blast_record = blast_records.next()
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
StopIteration
# No further records
或者,你也可以使用 for - 循环
>>> for blast_record in blast_records:
... # Do something with blast_record
注意对每个BLAST搜索结果只能迭代一次。通常,对于每个BLAST记录,你可能会保存你 感兴趣的信息。如果你想保存所有返回的BLAST记录,你可以把迭代 转换成列表。
>>> blast_records = list(blast_records)
现在,你可以像通常的做法通过索引从这个列表中获得每一条BLAST结果。 如果你的BLAST输出 结果文件很大,那么当把它们全部放入一个列表时,你也许会遇到内存不够的情况。
一般来说,你会一次运行一个BLAST搜索。然后,你只需提取第一条BLAST 搜索记录到
blast_records :
>>> from Bio.Blast import NCBIXML
>>> blast_records = NCBIXML.parse(result_handle)
>>> blast_record = blast_records.next()
or more elegantly:
或者更加优雅地:
>>> from Bio.Blast import NCBIXML
>>> blast_record = NCBIXML.read(result_handle)
我猜你现在在想BLAST搜索记录中到底有什么。
7.4 BLAST 记录类¶
一个BLAST搜索结果记录包括了所有你想要从中提取出来的信息。现在,我们将 用一个例子说明你怎么从BLAST搜索结果提取出一些信息。但是,如果你想从BLAST 搜索结果获得的信息没有在这里提到,你可以详细阅读BLAST搜索记录类, 并且可以参考下源代码 或者 是自动生成的文档 - 文档字符串里面包含了许多 关于各部分源代码是什么的很有用的信息。
继续我们的例子,让我们打印出所有大于某一特定阈值的BLAST命中结果的一些汇总信息。 代码如下:
>>> E_VALUE_THRESH = 0.04
>>> for alignment in blast_record.alignments:
... for hsp in alignment.hsps:
... if hsp.expect < E_VALUE_THRESH:
... print '****Alignment****'
... print 'sequence:', alignment.title
... print 'length:', alignment.length
... print 'e value:', hsp.expect
... print hsp.query[0:75] + '...'
... print hsp.match[0:75] + '...'
... print hsp.sbjct[0:75] + '...'
上面代码会打印出如下图的总结报告:
****Alignment****
sequence: >gb|AF283004.1|AF283004 Arabidopsis thaliana cold acclimation protein WCOR413-like protein
alpha form mRNA, complete cds
length: 783
e value: 0.034
tacttgttgatattggatcgaacaaactggagaaccaacatgctcacgtcacttttagtcccttacatattcctc...
||||||||| | ||||||||||| || |||| || || |||||||| |||||| | | |||||||| ||| ||...
tacttgttggtgttggatcgaaccaattggaagacgaatatgctcacatcacttctcattccttacatcttcttc...
基本上,一旦你解析了BLAST搜索结果文件,你可以提取任何你需要的信息。 当然,这取决于你想要获得什么信息。但是希望这里的例子能够帮助你开始工作。
在用Biopython提取BLAST搜索结果信息的时候,重要的是你需要考虑到信息存储在什么
(Biopython)对象中。在Biopython中,解析器返回 Record 对象,这个对象
可以是 Blast 类型的,也可以是 PSIBlast 类型的,具体哪个取决你
解析什么。这些对象的定义都可以在 Bio.Blast.Record 找到 并且很完整。
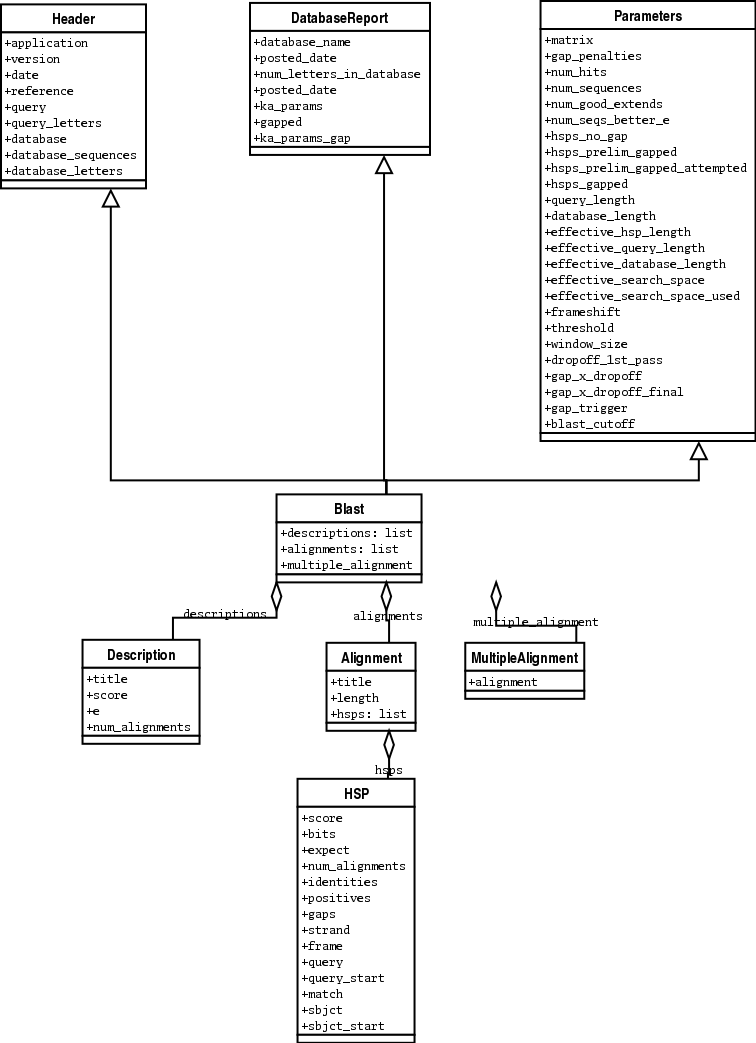
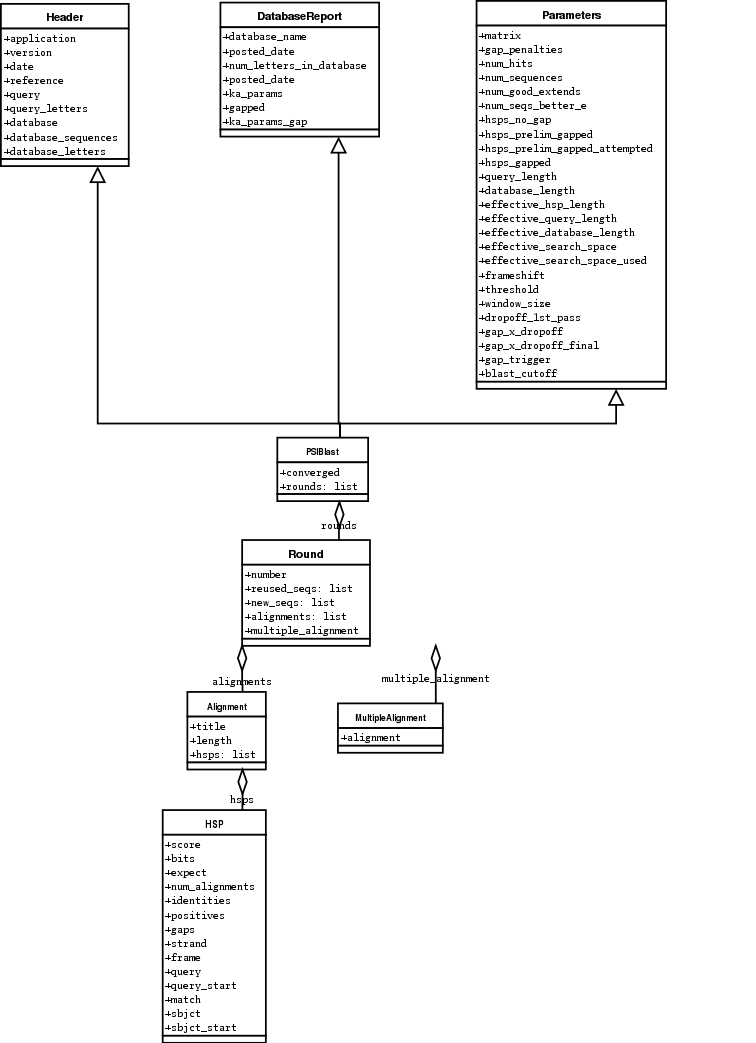
下面是 我尝试画的 Blast 和 PSIBlast 记录类的UML图。如果你对UML图很熟悉,不妨
看看下面的UML图是否有错误或者可以改进的地方,如果有,请联系我。
BLAST类图在这里 7.4 。
PSIBlast 记录类是类似的,但是支持用在迭代器中的rounds方法。PSIBlast类图在这里 7.4 。
7.5 废弃的BLAST 解析器¶
老版本的Biopython 有针对纯文本和HTML格式输出结果的解析器。但是经过几年 我们发现维护这些解析器很困难。基本上,任何BLAST输出的任何小改变都会导致 这些解析器失效。所以我们推荐你解析XML格式的BLAST输出结果,就像在 7.3 描述的那样。
取决于你使用Biopython的版本,纯文本格式的解析器也许有效也许失效。 用这个解析器的所带来的风险由你自己承担。
7.5.1 解析纯文本格式的BLAST输出¶
纯文本格式的解析器在 Bio.Blast.NCBIStandalone 。
和xml解析器类似, 我们也需要一个能够传给解析器的文件句柄。这个文件句柄必须
实现了 readline() 方法 。通常要获得这样文件句柄,既可以用Biopython提供的
blastall 或 blastpgp 函数来调用本地的BLAST,或者从命令行运行本地的
BLAST, 并且如下处理:
>>> result_handle = open("my_file_of_blast_output.txt")
好了,既然现在得到了个文件句柄(我们称它是 result_handle ),
我们已经做好了解析它的准备。按下面的代码来解析:
>>> from Bio.Blast import NCBIStandalone
>>> blast_parser = NCBIStandalone.BlastParser()
>>> blast_record = blast_parser.parse(result_handle)
这样就能把BALST的搜索结果报告解析到Blast记录类中(取决你于你解析的对象, 解析结果可能返回一条 Blast 或者 PSIBlast记录)。这样你就可以从中提取 信息了。在我们的例子里,我们来打印出大于某个阈值的所有比对的一个总结 信息。
>>> E_VALUE_THRESH = 0.04
>>> for alignment in blast_record.alignments:
... for hsp in alignment.hsps:
... if hsp.expect < E_VALUE_THRESH:
... print '****Alignment****'
... print 'sequence:', alignment.title
... print 'length:', alignment.length
... print 'e value:', hsp.expect
... print hsp.query[0:75] + '...'
... print hsp.match[0:75] + '...'
... print hsp.sbjct[0:75] + '...'
如果你已经读过 7.3 节关于解析XML格式的部分, 你将会发现上面的代码和那个章节的是一样的。一旦你把输出文件解析到记录类中, 你就能处理信息,不管你原来的BLAST输出格式是什么。很赞吧。
好,解析一条记录是不错,那么如果我有一个包含许多记录的BLAST文件 - 我该怎么处理它们呢?好吧,不要害怕,答案就在下个章节中。
7.5.2 解析包含多次BLAST结果的纯文本BLAST文件¶
我们可以用BLAST迭代器解析多次结果。为了得到一个迭代器,我们首先需要创建一个解析器,来 解析BLAST的搜索结果报告为Blast记录对象。
>>> from Bio.Blast import NCBIStandalone
>>> blast_parser = NCBIStandalone.BlastParser()
然后,我们假定我们有一个连接到一大堆blast记录的文件句柄,我们把这个文件句柄
叫做 result_handle 。 怎么得到一个文件句柄在上面blast解析章节有详细
描述。
好了,我们现在有了一个解析器和一个文件句柄,我们可以用以下命令来创建 一个迭代器。
>>> blast_iterator = NCBIStandalone.Iterator(result_handle, blast_parser)
第二个参数,解析器,是可选的。如果我们没有提供一个解析器,那么迭代器将会 一次返回一个原始的BLAST搜索结果。
现在我们已经有了个迭代器,就可以开始通过 next() 方法来获取BLAST
记录(由我们的解析器产生)。
>>> blast_record = blast_iterator.next()
每次调用next都会返回一条我们能处理的新记录。现在我们可以遍历所有记录,并打印一 个我们最爱、漂亮的、简洁的BLAST记录报告。
>>> for blast_record in blast_iterator:
... E_VALUE_THRESH = 0.04
... for alignment in blast_record.alignments:
... for hsp in alignment.hsps:
... if hsp.expect < E_VALUE_THRESH:
... print '****Alignment****'
... print 'sequence:', alignment.title
... print 'length:', alignment.length
... print 'e value:', hsp.expect
... if len(hsp.query) > 75:
... dots = '...'
... else:
... dots = ''
... print hsp.query[0:75] + dots
... print hsp.match[0:75] + dots
... print hsp.sbjct[0:75] + dots
迭代器允许你处理很多blast记录而不出现内存不足的问题。因为,它使一次处理 一个记录。我曾经用大处理过一个非常巨大的文件,没有出过任何问题。
7.5.3 在巨大的BLAST纯文本文件中发现不对的记录¶
当我开始解析一个巨大的blast 文件,有时候会碰到一个郁闷的问题就是解析器以一个 ValueError异常终止了。这是个严肃的问题。因为你无法分辨导致ValueError异常的是 解析器的问题还是BLAST的问题。更加糟糕是,你不知道在哪一行解析器失效了。所以, 你不能忽略这个错误。不然,可能会忽视一个重要的数据。
我们以前必须写一些小脚本来解决这个问题。不过,现在 Bio.Blast 模块包含了
BlastErrorParser ,可以更加简单地来解决这个问题。 BlastErrorParser
和常规的 BlastParser 类似,但是它加了特别一层来捕获由解析器产生的ValueErrors
异常,并尝试来诊断这些错误。
让我们来看看怎样用这个解析器 - 首先我们定义我们准备解析的文件和报告错误情况的 输出文件。
>>> import os
>>> blast_file = os.path.join(os.getcwd(), "blast_out", "big_blast.out")
>>> error_file = os.path.join(os.getcwd(), "blast_out", "big_blast.problems")
现在我们想要一个 BlastErrorParser :
>>> from Bio.Blast import NCBIStandalone
>>> error_handle = open(error_file, "w")
>>> blast_error_parser = NCBIStandalone.BlastErrorParser(error_handle)
注意,解析器有个关于文件句柄的可选参数。如果传递了这个参数,那么解析器就会 把产生ValueError异常的记录写到这个文件句柄中。不然的话,这些错误记录就不会 被记录下来。
现在,我们可以像用常规的blast解析器一样地用 BlastErrorParser 。
特别的是,我们也许想要一个一次读入一个记录的迭代器并用 BlastErrorParser
来解析它。
>>> result_handle = open(blast_file)
>>> iterator = NCBIStandalone.Iterator(result_handle, blast_error_parser)
我们可以一次读一个记录,并且我们现在可以捕获并处理那些因为Blast引起的、 不是解析器本身导致的错误。
>>> try:
... next_record = iterator.next()
... except NCBIStandalone.LowQualityBlastError, info:
... print "LowQualityBlastError detected in id %s" % info[1]
.next() 方法通常被 for 循环间接地调用。现在, BlastErrorParser
能够捕获如下的错误:
ValueError- 这就是和常规BlastParser产生的一样的错误。这个错误产生 是因为解析器不能解析某个文件。通常是因为解析器有bug, 或者是 因为你使用解析器的版本和你BLAST命令的版本不一致。LowQualityBlastError- 当Blast一条低质量的序列时(比如,一条 只有1个核苷酸的短序列),似乎Blast会终止并屏蔽掉整个序列,所有就没有什么可以 解析了。 这种情况下,Blast就会产生一个不完整的报告导致解析器出现ValueError 错误。LowQualityBlastError错误在这种情况下产生。这个错误返回如下 信息:item[0]– The error messageitem[0]- 错误消息item[1]– The id of the input record that caused the error. This is really useful if you want to record all of the records that are causing problems.item[1]- 导致错误产生的输入记录id。如果你想记录所有导致问题 记录的时候很有用。
就像上面提到的那样,BlastErrorParser 将会把有问题的记录写到指定的``error_handle``。 然后,你可以排查这些有问题记录。你可以针对某条记录来调试解析器,或者找到 你运行blast中的问题。无论哪种方式,这些都是有用的经验。
希望 BlastErrorParser 能帮你更简单的调试和处理一些数据巨大的Blast 文件。
7.6 处理PSI-BLAST¶
你可以通过 Bio.Blast.Applications 模块中的包装函数来运行单机版本的PSI-BLAST
(老版本的NCBI命令工具行 blastpgp 或者它的替代程序 psiblast )。
在写这篇指南的时候,没有迹象表明NCBI将会支持通过internet来进行PSI-BLAST 搜索。
请注意 Bio.Blast.NCBIXML 解析器能读入并解析当前版本PSI-BLAST的、XML格式的
输出,但是像哪条序列在每个迭代循环中是新的还是复用的信息在XML格式输出中是没有的。
如果,你需要这些信息你应该用纯文本输出和 Bio.Blast.NCBIStandalone 模块的
PSIBlastParser 。
7.7 处理 RPS-BLAST¶
你可以通过 Bio.Blast.Applications 模块中的包装函数来运行单机版本的RPS-BLAST
(或者老版本的NCBI命令工具行 rpsblast 或者同样名字的替代程序 )。
在写这篇指南的时候,没有迹象表明NCBI将会支持通过internet来进行RPS-BLAST 搜索
你可以通过 Bio.Blast.NCBIXML 这个解析器来读入并解析当前版本的RPS-BLAST的
XML格式的输出。