第12章 Bio.PopGen:群体遗传学¶
Bio.PopGen是一个群体遗传学相关的模块,在Biopython 1.44及以后的版本中可用。
该模块的中期目标是支持各种类型的数据格式、应用程序和数据库。目前,该模块正在紧张的开发中,并会快速实现对新特征的支持。这可能会带来一些不稳定的API,尤其是当你使用的是开发版。不过,我们正式公开发行的API应该更加稳定。
12.1 GenePop¶
GenePop( http://genepop.curtin.edu.au/)是一款主流的群体遗传学软件包,支持Hardy-Weinberg检验、连锁不平衡、群体分化、基础统计计算、 \(F_{st}\) 和迁移率估计等等。GenePop并不支持基于序列的统计计算,因为它并不能处理序列数据。GenePop文件格式广泛用于多种其它的群体遗传学应用软件,因此成为群体遗传学领域重要格式。
Bio.PopGen提供GenePop文件格式解析器和生成器,同时也提供操作记录内容的小工具。此处有个关于怎样读取GenePop文件的示例(你可以在Biopython的Test/PopGen文件夹下找到GenePop示例文件):
from Bio.PopGen import GenePop
handle = open("example.gen")
rec = GenePop.read(handle)
handle.close()
它将读取名为example.gen的文件并解析。如果你输出rec,那么该记录将会以GenePop格式再次输出。
在rec中最重要的信息是基因座名称和群体信息(当然不止这些,请使用help(GenePop.Record)获得API帮助文档)。基因座名称可以在rec.loci_list中找到,群体信息可以在rec.populations中找到。群体信息是一个列表,每个群体(population)作为其中一个元素。每个元素本身又是包含个体(individual)的列表,每个个体包含个体名和等位基因列表(每个marker两个元素),下面是一个rec.populations的示例:
[
[
('Ind1', [(1, 2), (3, 3), (200, 201)],
('Ind2', [(2, None), (3, 3), (None, None)],
],
[
('Other1', [(1, 1), (4, 3), (200, 200)],
]
]
在上面的例子中,我们有两个群体,第一个群体包含两个个体,第二个群体只包含一个个体。第一个群体的第一个个体叫做Ind1,紧接着是3个基因座各自的等位基因信息。请注意,对于任何的基因座,信息可以缺失(如上述个体Ind2)。
有几个可用的工具函数可以处理GenePop记录,如下例:
from Bio.PopGen import GenePop
#Imagine that you have loaded rec, as per the code snippet above...
rec.remove_population(pos)
#Removes a population from a record, pos is the population position in
# rec.populations, remember that it starts on position 0.
# rec is altered.
rec.remove_locus_by_position(pos)
#Removes a locus by its position, pos is the locus position in
# rec.loci_list, remember that it starts on position 0.
# rec is altered.
rec.remove_locus_by_name(name)
#Removes a locus by its name, name is the locus name as in
# rec.loci_list. If the name doesn't exist the function fails
# silently.
# rec is altered.
rec_loci = rec.split_in_loci()
#Splits a record in loci, that is, for each loci, it creates a new
# record, with a single loci and all populations.
# The result is returned in a dictionary, being each key the locus name.
# The value is the GenePop record.
# rec is not altered.
rec_pops = rec.split_in_pops(pop_names)
#Splits a record in populations, that is, for each population, it creates
# a new record, with a single population and all loci.
# The result is returned in a dictionary, being each key
# the population name. As population names are not available in GenePop,
# they are passed in array (pop_names).
# The value of each dictionary entry is the GenePop record.
# rec is not altered.
GenePop不支持群体名,这种限制有时会很麻烦。Biopython对群体名的支持正在规划中,这些功能扩展仍会保持对标准格式的兼容性。同时,中期目标是对GenePop网络服务的支持。
12.2 溯祖模拟(Coalescent simulation)¶
溯祖模拟是一种对群体遗传学信息根据时间向后推算的模型(backward model)。对祖先的模拟是通过寻找到最近共同祖先(Most Recent Common Ancestor,MRCA)完成。从MRCA到目前这一代样本间的血统关系有时称为家系(genealogy)。简单的情况是假定群体大小固定,单倍型,无群体结构,然后模拟无选择压的单个基因座的等位基因。
溯祖理论被广泛用于多种领域,如选择压力检测、真实群体的群体参数估计以及疾病基因图谱。
Biopython对溯祖的实现不是去创建一个新的内置模拟器,而是利用现有的SIMCOAL2( http://cmpg.unibe.ch/software/simcoal2/ )。与其他相比,SIMCOAL2允许存在群体结构,多群体事件,多种类型的可发生重组的基因座(SNPs,序列,STRs/微卫星和RFLPs),具有多染色体的二倍体和测量偏倚(ascertainment bias)。注意,SIMCOAL2并不支持所有的选择模型。建议阅读上述链接中的SIMCOAL2帮助文档。
SIMCOAL2的输入是一个指定所需的群体和基因组的文件,输出是一系列文件(通常在1000个左右),它们包括每个亚群(subpopulation)中模拟的个体基因组。这些文件有多种途径,如计算某些统计数据(e.g. \(F_{st}\) 或Tajima D)的置信区间以得到可信的范围。然后将真实的群体遗传数据统计结果与这些置信区间相比较。
Biopython溯祖模拟可以创建群体场景(demographic scenarios)和基因组,然后运行SIMCOAL2。
12.2.1 创建场景(scenario)¶
创建场景包括创建群体及其染色体结构。多数情况下(如计算近似贝斯估计量(Approximate Bayesian Computations – ABC)),测试不同参数的变化很重要(如不同的有效群体大小 \(N_e\) , 从10,50,500到1000个体)。提供的代码可以很容易地模拟具有不同群体参数的场景。
下面我们将学习怎样创建场景,然后是怎样进行模拟。
12.2.1.1 群体¶
有一些内置的预定义群体,均包含两个共同的参数:同群种(deme)的样本大小(在模板中称为 sample_size ,其使用请见下文)和同群种大小,如亚群大小(pop_size)。所有的群体都可以作为模板,所有的参数也可以变化,每个模板都有自己的系统名称。这些预定义的群体/模板(template)包括:
- Single population, constant size
- 单一种群固定群体大小。标准的参数即可满足它,模板名称:simple.
- Single population, bottleneck
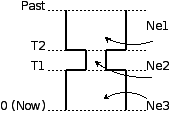
- 单一群体瓶颈效应,如图 12.2.1.1 所示。参数有当前种群大小(图中ne3模板的pop_size)、种群扩张时间 - 扩张发生后代数(expand_gen),瓶颈发生时的有效群体大小(ne2),种群收缩时间(contract_gen)以及原始种群大小(ne3)。模板名:bottle。
- Island model
- 典型的岛屿模型。同群种(deme)总数表示为total_demes,迁移率表示为mig。模板名:island。
- Stepping stone model - 1 dimension
- 一维脚踏石模型(Stepping stone model),极端状态下种群分布不连续。同群种(deme)总数表示为total_demes,迁移率表示为mig。模板名:ssm_1d。
- Stepping stone model - 2 dimensions
- 二维脚踏石模型,极端状态下种群分布不连续。参数有表示水平维度的x和表示垂直维度的y(同群种总数即为x × y),以及表示迁移率的mig。模板名:ssm_2d。
在我们的第一个示例中,将生成一个单一种群固定群体大小(Single population, constant size)模板,样本大小(sample size)为30,同群种大小(deme size)为500。代码如下:
from Bio.PopGen.SimCoal.Template import generate_simcoal_from_template
generate_simcoal_from_template('simple',
[(1, [('SNP', [24, 0.0005, 0.0])])],
[('sample_size', [30]),
('pop_size', [100])])
执行该段代码将会在当前目录生成一个名为simple_100_300.par的文件,该文件可作为SIMCOAL2的输入文件,用于模拟群体(下面将会展示Biopython是如何调用SIMCOAL2)。
这段代码仅包含一个函数的调用,让我们一个参数一个参数地讨论。
第一个参数是模板id(从上面的模板列表中选择)。我们使用 ’simple’,表示的是单一群体固定种群大小模板。
第二个参数是染色体结构,将在下一节详细阐述。
第三个参数是所有需要的参数列表及其所有可能的值(此列中所有的参数都只含有一个可能值)。
现在让我们看看岛屿模型示例。我们希望生成几个岛屿模型,并对不同大小的同群种感兴趣:10、50和100,迁移率为1%。样本大小和同群种大小与上一个示例一致,代码如下:
from Bio.PopGen.SimCoal.Template import generate_simcoal_from_template
generate_simcoal_from_template('island',
[(1, [('SNP', [24, 0.0005, 0.0])])],
[('sample_size', [30]),
('pop_size', [100]),
('mig', [0.01]),
('total_demes', [10, 50, 100])])
此例将会生成3个文件:island_100_0.01_100_30.par,island_10_0.01_100_30.par 和 island_50_0.01_100_30.par。注意,生成文件名的规律是:模板名,然后是参数值逆序排列。
还有一些存在较多争议的群体模板(请见Biopython源代码中Bio/PopGen/SimCoal/data文件夹)。同时,用户可以创建新的模板,该功能将在以后的文档中讨论。
12.2.1.2 染色体结构¶
我们强烈建议你阅读SIMCOAL2文档,以完整理解染色体结构建模的各种使用。在本小节,我们只讨论如何使用Biopython接口实现指定的染色体结构,不会涉及SIMCOAL2可实现哪些染色体结构。
我们首先实现一条染色体,包含24个SNPs,每个相邻基因座的重组率为0.0005,次等位基因的最小频率为0。这些由以下列表指定(作为第二个参数传递给generate_simcoal_from_template函数):
[(1, [('SNP', [24, 0.0005, 0.0])])]
这实际上是上一个示例使用的染色体结构。
染色体结构表示为一个包含所有染色体的列表,每条染色体(即列表中的每个元素)由一个元组(tuple)组成,元组包括一对元素组成。元组的第一个元素是染色体被重复的次数(因为有可能需要多次重复同一条染色体)。元组的第二个元素是一个表示该染色体的实际组成的列表,每个列表元素又包括一对元素,第一个是基因座类型,第二个是该基因座的参数列表。是否有点混淆了呢?在我们展示示例之前,先让我们回顾下上一个示例:我们有一个列表(表示一条染色体),该染色体只有一个实例(因此不会被重复),它由24个SNPs组成,每个相邻SNP间的重组率为0.0005,次等位基因的最小频率为0.0(即它可以在某些染色体中缺失)。
现在让我们看看更复杂的示例:
[
(5, [
('SNP', [24, 0.0005, 0.0])
]
),
(2, [
('DNA', [10, 0.0, 0.00005, 0.33]),
('RFLP', [1, 0.0, 0.0001]),
('MICROSAT', [1, 0.0, 0.001, 0.0, 0.0])
]
)
]
首先,我们有5条与上一示例具有相同结构组成的染色体(即24SNPs)。然后是2条这样的染色体:包含一段具有重组率为0.0、突变率为0.0005及置换率为0.33的10个核苷酸长度的DNA序列,一段具有重组率为0.0、突变率为0.0001的RFLP,一段具有重组率为0.0、突变率为0.001、几何参数为0.0、范围限制参数为0.0的微卫星(microsatellite,STR)序列(注意,因为这是单个微卫星,接下来没有基因座,因此这里的重组率没有任何影响,更多关于这些参数的信息请查阅SIMCOAL2文档,你可以使用它们模拟各种突变模型,包括典型的微卫星渐变突变模型)。
12.2.2 运行SIMCOAL2¶
现在我们讨论如何从Biopython内部运行SIMCOAL2。这需要SIMCOAL2的可执行二进制文件名为simcoal2(在Windows平台下为simcoal2.exe),请注意,从官网下载的程序命名格式通常为simcoal2_x_y。因此,当安装SIMCOAL2时,需要重命名可执行文件,这样Biopython才能正确调用。
SIMCOAL2可以处理不是使用上诉方法生成的文件(如手动配置的参数文件),但是我们将使用上述方法得到的文件创建模型:
from Bio.PopGen.SimCoal.Template import generate_simcoal_from_template
from Bio.PopGen.SimCoal.Controller import SimCoalController
generate_simcoal_from_template('simple',
[
(5, [
('SNP', [24, 0.0005, 0.0])
]
),
(2, [
('DNA', [10, 0.0, 0.00005, 0.33]),
('RFLP', [1, 0.0, 0.0001]),
('MICROSAT', [1, 0.0, 0.001, 0.0, 0.0])
]
)
],
[('sample_size', [30]),
('pop_size', [100])])
ctrl = SimCoalController('.')
ctrl.run_simcoal('simple_100_30.par', 50)
需要注意的是最后两行(以及新增的import行)。首先是创建一个应用程序控制器对象,需要指定二进制可执行文件所在路径。
模拟器在最后一行运行:从上述阐述的规律可知,文件名为simple_100_30.par的输入文件是我们创建的模拟参数文件,然后我们指定了希望运行50次独立模拟。默认情况下,Biopython模拟二倍体数据,但是可以添加第三个参数用于模拟单倍体数据(字符串’0’)。然后,SIMCOAL2将会执行(这需要运行很长时间),并创建一个包含模拟结果的文件夹,结果文件可便可用于分析(尤其是研究Arlequin3数据)。在未来的Biopython版本中,可能会支持Arlequin3格式文件的读取,从而在Biopython中便能分析SIMCOAL2结果。
12.3 其它应用程序¶
这里我们讨论一些处理其它的群体遗传学中应用程序的接口和小工具,这些应用程序具有争议,使用得较少。
12.3.1 FDist:检测选择压力和分子适应¶
FDist是一个选择压力检测的应用程序包,基于通过 \(F_{st}\) 和杂合度计算(即模拟)得到的“中性”(“neutral”)置信区间。“中性”置信区间外的Markers(可以是SNPs,微卫星,AFLPs等等)可以被认为是候选的受选择marker。
FDist主要运用在当marker数量足够用于估计平均 \(F_{st}\) ,而不足以从数据集中计算出离群点 - 直接地或者在知道大多数marker在基因组中的相对位置的情况下使用基于如Extended Haplotype Heterozygosity (EHH)的方法。
典型的FDist的使用如下:
- 从其它格式读取数据为FDist格式;
- 计算平均 \(F_{st}\) ,由FDist的datacal完成;
- 根据平均 \(F_{st}\) 和期望的总群体数模拟“中性”markers,这是核心部分,由FDist的fdist完成;
- 根据指定的置信范围(通常是95%或者是99%)计算置信区间,由cplot完成,主要用于对区间作图;
- 用模拟的“中性”置信区间评估每个Marker的状态,由pv完成,用于检测每个marker与模拟的相比的离群状态;
我们将以示例代码讨论每一步(FDist可执行二进制文件需要在PATH环境变量中)。
FDist数据格式是该应用程序特有的,不被其它应用程序使用。因此你需要转化你的数据格式到FDist可使用的格式。Biopython可以帮助你完成这个过程。这里有一个将GenePop格式转换为FDist格式的示例(同时包括后面示例将用到的import语句):
from Bio.PopGen import GenePop
from Bio.PopGen import FDist
from Bio.PopGen.FDist import Controller
from Bio.PopGen.FDist.Utils import convert_genepop_to_fdist
gp_rec = GenePop.read(open("example.gen"))
fd_rec = convert_genepop_to_fdist(gp_rec)
in_file = open("infile", "w")
in_file.write(str(fd_rec))
in_file.close()
在该段代码中,我们解析GenePop文件并转化为FDist记录(record)。
输出FDist记录将得到可以直接保存到可用于FDist的文件的字符串。FDist需要输入文件名为infile,因此我们将记录保存到文件名为infile的文件。
FDist记录最重要的字段(field)是:num_pops,群体数量;num_loci,基因座数量和loci_data,marker数据。记录的许多信息对用户来说可能没有用处,仅用于传递给FDist。
下一步是计算平均数据集的 \(F_{st}\) (以及样本大小):
ctrl = Controller.FDistController()
fst, samp_size = ctrl.run_datacal()
第一行我们创建了一个控制调用FDist软件包的对象,该对象被用于调用该包的其它应用程序。
第二行我们调用datacal应用程序,它用于计算 \(F_{st}\) 和样本大小。值得注意的是,用datacal计算得到的 \(F_{st}\) 是 Weir-Cockerham θ的“变种”( variation )。
现在我们可以调用主程序fdist模拟中性Markers。
sim_fst = ctrl.run_fdist(npops = 15, nsamples = fd_rec.num_pops, fst = fst,
sample_size = samp_size, mut = 0, num_sims = 40000)
- npops
- 现存自然群体数量,完全是一个“瞎猜值”(“guestimate”),必须小于100。
- nsamples
- 抽样群体数量,需要小于npops。
- fst
- 平均 \(F_{st}\) 。
- sample_size
- 每个群体抽样个体平均数
- mut
- 突变模型:0 - 无限等位基因突变模型;1 - 渐变突变模型
- num_sims
- 执行模拟的次数。通常,40000左右的数值即可,但是如果得到的执行区间范围比较大(可以通过下面的置信区间作图检测到),可以上调此值(建议每次调整10000次模拟)。
样本数量和样本大小措辞上的混乱源于原始的应用程序。
将会得到一个名为out.dat的文件,它包含模拟的杂合度和 \(F_{st}\) 值,行数与模拟的次数相同。
注意,fdist返回它可以模拟的平均 \(F_{st}\) ,关于此问题更多的细节,请阅读下面的“估计期望的平均 \(F_{st}\) ”
下一步(可选步骤)是计算置信区间:
cpl_interval = ctrl.run_cplot(ci=0.99)
只能在运行fdist之后才能调用cplot。
这将计算先前fdist结果的置信区间(此例中为99%)。第一个元素是杂合度,第二个是该杂合度的 \(F_{st}\) 置信下限,第三个是 \(F_{st}\) 平均值,第四个是置信上限。可以用于记录置信区间等高线。该列表也可以输出到out.cpl文件。
这步的主要目的是返回一系列的点用于对置信区间作图。如果只是需要根据模拟结果对每个marker的状态进行评估,可以跳过此步。
pv_data = ctrl.run_pv()
只能在运行fdist之后才能调用pv。
这将使用模拟marker对每个个体真实的marker进行评估,并返回一个列表,顺序与FDist记录中loci_list一致(loci_list又与GenePop顺序一致)。列表中每个元素包含四个元素,其中最重要的是最后一个元素(关于其他的元素,为了简单起见,我们不在这里讨论,请见pv帮助文档),它返回模拟的 \(F_{st}\) 低于 marker \(F_{st}\) 的概率。较大值说明极有可能是正选择(positive selection)marker,较小值表明可能是平衡选择(balancing selection)marker,中间值则可能是中性marker。怎样的值是“较大值”、“较小值”或者“中间值”是一个很主观的问题,但当使用置信区间方法及95%的置信区间时,“较大值”在0.95 - 1.00之间,“较小值”在0.00 - 0.05之间,“中间值”在0.05 - 0.05之间。
12.3.1.1 估计期望的平均 \(F_{st}\)¶
FDist通过对由下例公式得到的迁移率进行溯祖模拟估计期望的平均 \(F_{st}\) :
该公式有一些前提,比如种群大小无限大。
在实践中,当群体数量比较小,突变模型为渐进突变模型,样本大小增加,fdist将不能模拟得到可接受的近似平均 \(F_{st}\) 。
为了解决这个问题,Biopython提供了一个使用迭代方法的函数,通过依次运行几个fdist得到期望的值。该方法比运行单个fdist相比耗费更多计算资源,但是可以得到更好的结果。以下代码运行fdist得到期望的 \(F_{st}\) :
sim_fst = ctrl.run_fdist_force_fst(npops = 15, nsamples = fd_rec.num_pops,
fst = fst, sample_size = samp_size, mut = 0, num_sims = 40000,
limit = 0.05)
与run_fdist相比,唯一一个新的可选参数是limit,它表示期望的最大错误率。run_fdist可以(或许应该)由run_fdist_force_fst替代。
12.3.1.2 说明¶
计算平均 \(F_{st}\) 的过程可能比这里呈现的要复杂得多。更多的信息请查阅FDist README文件。同时,Biopython的代码也可用于实现更复杂的过程。
12.4 未来发展¶
最期望的是您的参与!
尽管如此,已经完成的功能模块正在逐步加入到Bio.PopGen,这些代码覆盖了程序FDist和SimCoal2,HapMap和UCSC Table Browser数据库,以及一些简单的统计计算,如 \(F_{st}\) , 或等位基因数。