第13章 Bio.Phylo系统发育分析¶
Biopython1.54开始引入了Bio.Phylo模块,与SeqIO和AlignIO类似,它的目的是提供 一个通用的独立于源数据格式的方法来使用系统进化树,同时提供一致的API来进行 I/O操作。
Bio.Phylo在一篇开放获取的期刊文章中有介绍 [9, Talevich et al., 2012], 这可能对您也有所帮助。
13.1 示例: 树中有什么?¶
为了熟悉这个模块,让我们首先从一个已经创建好的树开始,从几个不同的角度来审视 它。接着我们将给树的分支上颜色,并使用一个特殊的phyloXML特性,最终保存树。
译者注:本翻译中, 分支 对应原文中的 branch ,原文中一般代表 某一个节点的前导连线;而 进化枝 对应原文中的 clade ,代表某个节点所代表的整个 进化分支,包括本身和它所有的后代;若clade代表biopython中的对象则保留原文
在终端中使用你喜欢的编辑器创建一个简单的Newick文件:
% cat > simple.dnd <<EOF
> (((A,B),(C,D)),(E,F,G));
> EOF
这棵树没有分支长度,只有一个拓扑结构和标记的端点。(如果你有一个真正的树文件, 你也可以使用它来替代进行示例操作。)
选择启动你的Python解释器:
% ipython -pylab
对于交互式操作,使用参数 -pylab 启动IPython解释器能启用 matplotlib 整合
功能,这样图像就能自动弹出来。我们将在这个示例中使用该功能。
现在,在Python终端中,读取树文件,给定文件名和格式名。
>>> from Bio import Phylo
>>> tree = Phylo.read("simple.dnd", "newick")
以字符串打印该树对象我们将得到整个对象的层次结构概况。
>>> print tree
Tree(weight=1.0, rooted=False, name="")
Clade(branch_length=1.0)
Clade(branch_length=1.0)
Clade(branch_length=1.0)
Clade(branch_length=1.0, name="A")
Clade(branch_length=1.0, name="B")
Clade(branch_length=1.0)
Clade(branch_length=1.0, name="C")
Clade(branch_length=1.0, name="D")
Clade(branch_length=1.0)
Clade(branch_length=1.0, name="E")
Clade(branch_length=1.0, name="F")
Clade(branch_length=1.0, name="G")
Tree 对象包含树的全局信息,如树是有根树还是无根树。它包含一个根进化枝,
和以此往下以列表嵌套的所有进化枝,直至叶子分支。
函数 draw_ascii 创建一个简单的ASCII-art(纯文本)系统发生图。在没有更好
图形工具的情况下,这对于交互研究来说是一个方便的可视化展示方式。
>>> Phylo.draw_ascii(tree)
________________________ A
________________________|
| |________________________ B
________________________|
| | ________________________ C
| |________________________|
_| |________________________ D
|
| ________________________ E
| |
|________________________|________________________ F
|
|________________________ G
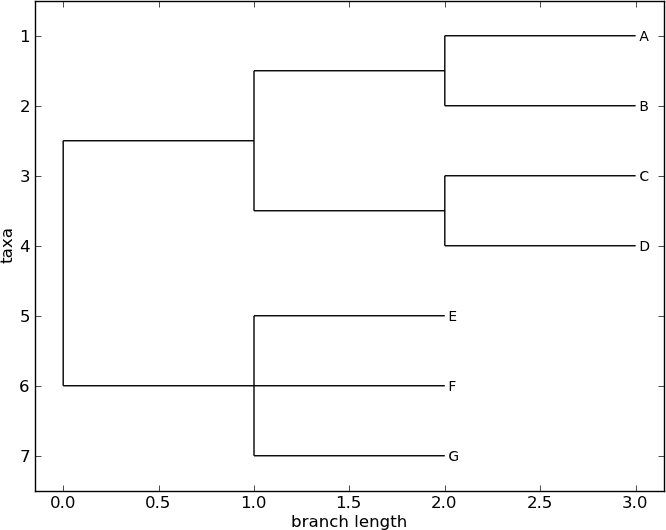
如果你安装有 matplotlib 或者 pylab, 你可以使用 draw 函数一个图像(见 Fig.
13.1 ):
>>> tree.rooted = True
>>> Phylo.draw(tree)
13.1.1 给树的分支上颜色¶
函数 draw 和 draw_graphviz 支持在树中显示不同的颜色和分支宽度。
从Biopython 1.59开始,Clade对象就开始支持 color 和 width 属性,
且使用他们不需要额外支持。这两个属性都表示导向给定的进化枝前面的分支的
属性,并依次往下作用,所以所有的后代分支在显示时也都继承相同的宽度和颜
色。
在早期的Biopython版本中,PhyloXML树有些特殊的特性,使用这些属性需要首先 将这个树转换为一个基本树对象的子类Phylogeny,该类在Bio.Phylo.PhyloXML模 块中。
在Biopython 1.55和之后的版本中,这是一个很方便的树方法:
>>> tree = tree.as_phyloxml()
在Biopython 1.54中, 你能通过导入一个额外的模块实现相同的事情:
>>> from Bio.Phylo.PhyloXML import Phylogeny
>>> tree = Phylogeny.from_tree(tree)
注意Newick和Nexus文件类型并不支持分支颜色和宽度,如果你在Bio.Phylo中使用 这些属性,你只能保存这些值到PhyloXML格式中。(你也可以保存成Newick或Nexus 格式,但是颜色和宽度信息在输出的文件时会被忽略掉。)
现在我们开始指定颜色。首先,我们将设置根进化枝为灰色。我们能通过赋值24位 的颜色值来实现,用三位数的RGB值、HTML格式的十六进制字符串、或者预先设置好的 颜色名称。
>>> tree.root.color = (128, 128, 128)
Or:
>>> tree.root.color = "#808080"
Or:
>>> tree.root.color = "gray"
一个进化枝的颜色会被当作从上而下整个进化枝的颜色,所以我们这里设置根的 的颜色会将整个树的颜色变为灰色。我们能通过在树中下面分支赋值不同的颜色 来重新定义某个分支的颜色。
让我们先定位“E”和“F”最近祖先(MRCA)节点。方法 common_ancestor 返回
原始树中这个进化枝的引用,所以当我们设置该进化枝为“salmon”颜色时,这个颜
色则会在原始的树中显示出来。
>>> mrca = tree.common_ancestor({"name": "E"}, {"name": "F"})
>>> mrca.color = "salmon"
当我们碰巧明确地知道某个进化枝在树中的位置,以嵌套列表的形式,我们就能
通过索引的方式直接跳到那个位置。这里,索引 [0,1] 表示根节点的第一个
子代节点的第二个子代。
>>> tree.clade[0,1].color = "blue"
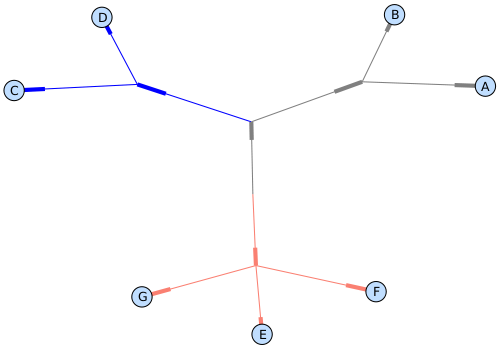
最后,展示一下我们的工作结果 (见 Fig. 13.1.1 ):
>>> Phylo.draw(tree)

注意进化枝的颜色包括导向它的分支和它的子代的分支。E和F的共同祖先结果刚好 在根分支下面,而通过这样上色,我们能清楚的看出这个树的根在哪里。
我们已经完成了很多!现在让我们休息一下,保存一下我们的工作。使用一个文件
名或句柄(这里我们使用标准输出来查看将会输出什么)和 phyloxml 格式来
调用 write 函数。PhyloXML格式保存了我们设置的颜色,所以你能通过其他树
查看工具,如Archaeopteryx,打开这个phyloXML文件,这些颜色也会显示出来。
>>> import sys
>>> Phylo.write(tree, sys.stdout, "phyloxml")
<phy:phyloxml xmlns:phy="http://www.phyloxml.org">
<phy:phylogeny rooted="true">
<phy:clade>
<phy:branch_length>1.0</phy:branch_length>
<phy:color>
<phy:red>128</phy:red>
<phy:green>128</phy:green>
<phy:blue>128</phy:blue>
</phy:color>
<phy:clade>
<phy:branch_length>1.0</phy:branch_length>
<phy:clade>
<phy:branch_length>1.0</phy:branch_length>
<phy:clade>
<phy:name>A</phy:name>
...
本章的其余部分将更加细致的介绍Bio.Phylo核心功能。关于Bio.Phylo的更多例 子,请参见Biopython.org上的Cookbook手册页面。
13.2 I/O 函数¶
和SeqIO、AlignIO类似, Phylo使用四个函数处理文件的输入输出: parse 、
read 、 write 和 convert ,所有的函数都支持Newick、NEXUS、
phyloXML和NeXML等树文件格式。
read 函数解析并返回给定文件中的单个树。注意,如果文件中包含多个或不包含任何树,它将抛出一个错误。
>>> from Bio import Phylo
>>> tree = Phylo.read("Tests/Nexus/int_node_labels.nwk", "newick")
>>> print tree
(Biopython发布包的 Tests/Nexus/ 和 Tests/PhyloXML/ 文件夹中有相应的例子)
处理多个(或者未知个数)的树文件,需要使用 parse 函数迭代给定文件中的每一个树。
>>> trees = Phylo.parse("Tests/PhyloXML/phyloxml_examples.xml", "phyloxml")
>>> for tree in trees:
... print tree
使用 write 函数输出一个或多个可迭代的树。
>>> trees = list(Phylo.parse("phyloxml_examples.xml", "phyloxml"))
>>> tree1 = trees[0]
>>> others = trees[1:]
>>> Phylo.write(tree1, "tree1.xml", "phyloxml")
1
>>> Phylo.write(others, "other_trees.xml", "phyloxml")
12
使用 convert 函数转换任何支持的树格式。
>>> Phylo.convert("tree1.dnd", "newick", "tree1.xml", "nexml")
1
>>> Phylo.convert("other_trees.xml", "phyloxml", "other_trees.nex", 'nexus")
12
和SeqIO和AlignIO类似,当使用字符串而不是文件作为输入输出时,需要使用 ‵‵StringIO`` 函数。
>>> from Bio import Phylo
>>> from StringIO import StringIO
>>> handle = StringIO("(((A,B),(C,D)),(E,F,G));")
>>> tree = Phylo.read(handle, "newick")
13.3 查看和导出树¶
了解一个 Tree 对象概况的最简单的方法是用 print 函数将它打印出来:
>>> tree = Phylo.read("Tests/PhyloXML/example.xml", "phyloxml")
>>> print tree
Phylogeny(rooted='True', description='phyloXML allows to use either a "branch_length"
attribute...', name='example from Prof. Joe Felsenstein's book "Inferring Phyl...')
Clade()
Clade(branch_length='0.06')
Clade(branch_length='0.102', name='A')
Clade(branch_length='0.23', name='B')
Clade(branch_length='0.4', name='C')
上面实际上是Biopython的树对象层次结构的一个概况。然而更可能的情况是,你希望见到 画出树的形状,这里有三个函数来做这件事情。
如我们在demo中看到的一样, draw_ascii 打印一个树的ascii-art图像(有根进化树)
到标准输出,或者一个打开的文件句柄,若有提供。不是所有关于树的信息被显示出来,但是它提供了一个
不依靠于任何外部依赖的快速查看树的方法。
>>> tree = Phylo.read("example.xml", "phyloxml")
>>> Phylo.draw_ascii(tree)
__________________ A
__________|
_| |___________________________________________ B
|
|___________________________________________________________________________ C
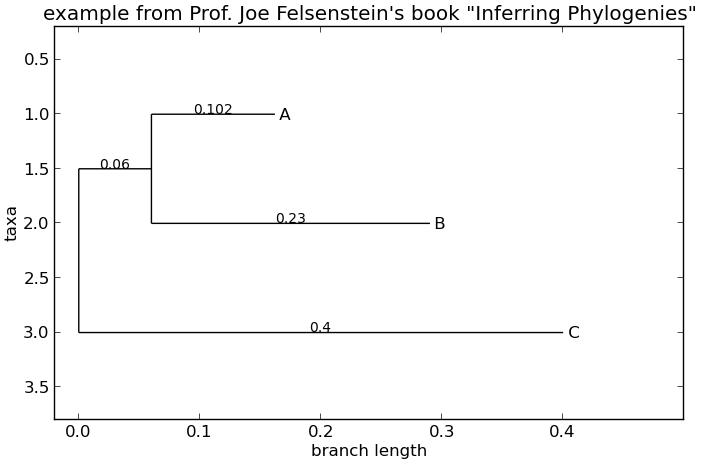
draw 函数则使用matplotlib类库画出一个更加好看的图像。查看API文档以获得关于它所接受的
用来定制输出的参数。
>>> tree = Phylo.read("example.xml", "phyloxml")
>>> Phylo.draw(tree, branch_labels=lambda c: c.branch_length)
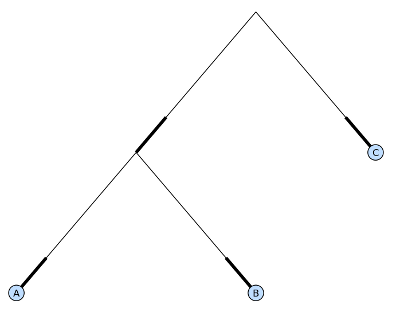
draw_graphviz 则画出一个无根的进化分枝图(cladogram),但是它要求你安装有Graphviz、
PyDot或PyGraphviz、Network和matplotlib(或pylab)。使用上面相同的例子,和Graphviz中的
dot 程序,让我们来画一个有根树(见图. 13.3 ):
>>> tree = Phylo.read("example.xml", "phyloxml")
>>> Phylo.draw_graphviz(tree, prog='dot')
>>> import pylab
>>> pylab.show() # Displays the tree in an interactive viewer
>>> pylab.savefig('phylo-dot.png') # Creates a PNG file of the same graphic
(提示:如果你使用 -pylab 选项执行IPython,调用 draw_graphviz 将导致matplotlib
查看器自动运行,而不需要手动的调用 show() 方法。)
这将输出树对象到一个NetworkX图中,使用Graphviz来布局节点的位置,并使用matplotlib来显示
它。这里有几个关键词参数来修改结果图像,包括大多数被NetworkX函数 networkx.draw 和
networkx.draw_graphviz 所接受的参数。
最终的显示也受所提供的树对象的 rooted 属性的影响。有根树在每个分支(branch)上显示
一个“head”来表明它的方向(见图. 13.3 ):
>>> tree = Phylo.read("simple.dnd", "newick")
>>> tree.rooted = True
>>> Phylo.draw_graphiz(tree)
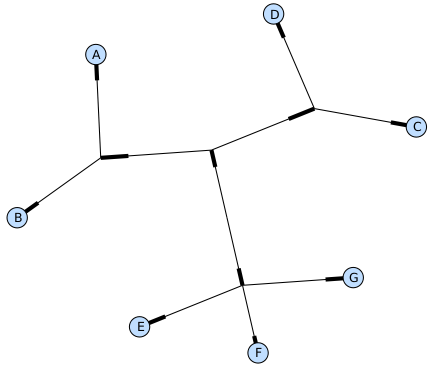
“prog”参数指定Graphviz的用来布局的引擎。默认的引擎 twopi 对任何大小的树都表现很好,
很可靠的避免交叉的分支出现。neato 程序可能画出更加好看的中等大小的树,但是有时候会
有交叉分支出现(见图. 13.3 )。 dot 程序或许对小型的树有用,
但是对于大一点的树的布局易产生奇怪的事情。
>>> Phylo.draw_graphviz(tree, prog="neato")
这个查看方式非常方便研究大型的树,因为matplotlib查看器可以放大选择的区域,使得杂乱的图像 变得稀疏。
>>> tree = Phylo.read("apaf.xml", "phyloxml")
>>> Phylo.draw_graphviz(tree, prog="neato", node_size=0)
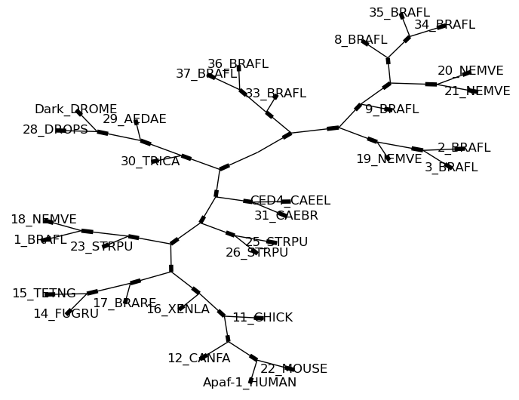
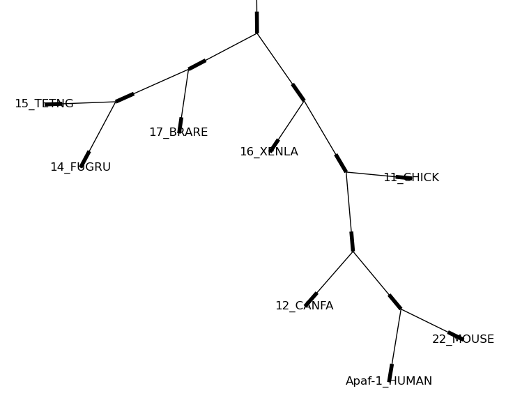
注意,分支长度并没有被正确地显示,因为Graphviz在布局时忽略了他们。然而,分支长度可以在输出
树为NetworkX图对象( to_networkx )时重新获得。
查看Biopython维基的Phylo页面
(http://biopython.org/wiki/Phylo)
以获得关于 draw_ascii 、 draw_graphviz 和 to_networkx 的更加高级的功能的描述
和例子。
13.4 使用Tree和Clade对象¶
parse 和 read 方法产生的 Tree 对象是一些包含递归的子树的容器,连接到 Tree
对象的 root 属性(不管进化树实际上被认为是否有根)。一个 Tree 包含进化树的全局信息,
如有根性(rootedness)和指向一个单独的 Clade 的引用; 一个 Clade 包含节点和进化枝
特异性信息,如分支长度(branch length)和一个它自身后代 Clade 实例的列表,附着在 clades
属性上。
所以,这里 tree 和 tree.root 间是有区别的. 然而,实际操作中,你几乎不需要担心它。为了
缓和这个不同,Tree 和 Clade 两者都继承自 TreeMixin,它包含常用的用来查找、审视和
修改树和任何它的进化枝的方法的实现。这意味着,所有 tree 所支持的方法在 tree.root 和
任何它下面的clade中都能用。( Clade 也有一个 root 属性,它返回clade对象本身。)
13.4.1 查找和遍历类方法¶
为了方便起见,我们提供了两个简化的方法来直接返回所有的外部或内部节点为列表:
- ``get_terminals``
- 创建一个包含树的所有末端(叶子)节点的列表。
- ``get_nonterminals``
- 创建一个包含树的所有非末端(内部)节点的列表。
这两个都包装了一个能完全控制树的遍历的方法 find_clades。另外两个遍历方法 find_elements
和 find_any 依赖于同样的核心功能,也接受同样的参数,没有更好的描述我们就把这个参数叫做
“目标说明”(target specification)吧。它们指定哪些树中的对象将被匹配并在迭代过程中返回。
第一个参数可以是下面的任何类型:
一个 TreeElement 实例 ,那个树的元素将根据一致性被匹配——这样,使用Clade实例作为目标将找到 树中的这个Clade;
一个 string ,匹配树元素的字符串表示——特别地,Clade的
name(在Biopython 1.56中引入);一个 class 或 type,这样每一个类型(或子类型)相同的树元素都被匹配;
一个 dictionary ,其中键(key)是树元素的属性名,值(value)将匹配到每个树元素相应的属性值。 它变得更加详细:
- 如果提供的是
int类型,它将匹配数值上相等的属性,即,1将匹配1或者1.0 - 如果提供的是boolean类型(True或者False),对应的属性值将被当做boolean求值和检验
None匹配None- 如果提供的是字符串,将被当做正则表达式对待(必须匹配对应元素属性的全部,不能只是前面的部分)。
提供没有特殊正则表达式字符的字符串将精准的匹配字符串属性,所以如果你不适用正则表达式,不用
担心它。例如,包含进化枝名称Foo1、Foo2和Foo3的一个树,
tree.find_clades({"name": "Foo1"})将匹配 Foo1,{"name": "Foo.*"}匹配所有的三个进化枝,而{"name": "Foo"}并不匹配任何进化枝。
由于浮点数值可能产生奇怪的行为,我们不支持直接匹配
floats 类型。作为替代,使用boolean值True来匹配每个元素中指定属性的非零值,然后再对这个属性用不等式(或精确地数值,如果你喜欢 危险地活着)进行手动过滤。如果该字典包含多个条目,匹配的元素必须匹配所有给定的属性值——以“and”方式思考,而不是“or”。
- 如果提供的是
一个接受一个参数(它将应用于树中的每一个元素),返回True或False的函数 function 。为方便起见, LookupError、AttributeError和ValueError被沉默,这样就提供了另外一个在树中查找浮点值的安全方式, 或者一些更加复杂的特性。
在目标参数后面,有两个可选的关键词参数:
- terminal
- — 用来选择或排除末端进化枝(或者叫叶子节点)的一个boolean值:True仅搜索末端进化枝,False则搜索
非末端(内部)进化枝,而默认为None,同时搜索末端和非末端进化枝,包括没有
is_terminal方法的 任何树元素。 - order
- — 树遍历的顺序:
"preorder"(默认值)是深度优先搜索(depth-first search,DFS),"postorder"是子节点先于父节点的DFS搜索,"level"是宽度优先搜索(breadth-first search,BFS)。
最后,这些方法接受任意的关键词参数,这些参数将被以和词典“目标说明”相同的方式对待:键表示要搜索的元素
属性的名称,参数值(string、integer、None或者boolean)将和找到的每个属性的值进行比较。如果没有提供
关键词参数,则任何TreeElement类型将被匹配。这个的代码普遍比传入一个词典作为“目标说明”要短:
tree.find_clades({"name": "Foo1"}) 可以简化为 tree.find_clades(name="Foo1")。
(在Biopython 1.56和以后的版本中,这可以更短:tree.find_clades("Foo1") )
现在我们已经掌握了“目标说明”,这里有一些遍历树的方法:
- ``find_clades``
查找每个包含匹配元素的进化枝。就是说,用
find_elements查找每个元素,然而返回对应的clade对象。 (这通常是你想要的。)最终的结果是一个包含所有匹配对象的迭代器,默认为深度优先搜索。这不一定是和Newick、Nexus或XML原文件 中显示的相同的顺序。
- ``find_elements``
- 查找和给定属性匹配的所有树元素,返回匹配的元素本身。简单的Newick树没有复杂的子元素,所以它将和
find_clades的行为一致。PhyloXML树通常在clade上附加有复杂的对象,所以这个方法对提取这些信息 非常有用。
- ``find_any``
- 返回
find_elements()所找到的第一个元素,或者None。这对于检测树中是否存在匹配的元素也非常有用, 可以在条件判断语句中使用。
另外两个用于帮助在树的节点间导航的方法:
- ``get_path``
- 直接列出从树的根节点(或当前进化枝)到给定的目标间的所有clade。返回包含这个路径上所有clade对象的 列表,以给定目标为结尾,但不包含根进化枝。
- ``trace``
- 列出树中两个目标间的所有clade对象,不包含起始和结尾。
13.4.2 信息类方法¶
这些方法提供关于整个树(或任何进化枝)的信息。
- ``common_ancestor``
- 查找所提供的所有目标的最近共同祖先(the most recent common ancestor) (这将是一个Clade对象)。如果没有提供任何目标,将返回当前Clade(调用该 方法的那个)的根;如果提供一个目标,将返回目标本身。然而,如果有任何提供 的目标无法在当前tree(或clade)中找到,将引起一个异常。
- ``count_terminals``
- 计算树中末端(叶子)节点的个数。
- ``depths``
- 创建一个树中进化枝到其深度的映射。结果是一个字典,其中键是树中所有的Clade
实例,值是从根到每个clade(包含末端)的距离。默认距离是到这个clade的分支
长度累加,然而使用
unit_branch_lengths=True选项,将只计算分支的个数 (其在树中的级数)。 - ``distance``
- 计算两个目标间的分支长度总和。如果只指定一个目标,另一个则为该树的根。
- ``total_branch_length``
- 计算这个树中的分支长度总和。这在系统发生学中通常就称为树的长度“length”, 但是我们使用更加明确的名称,以避免和Python的术语混淆。
余下的方法是boolean检测方法:
- ``is_bifurcating``
- 如果树是严格的二叉树;即,所有的节点有2个或者0个子代(对应的,内部或外部)。 根节点可能有三个后代,然而仍然被认为是二叉树的一部分。
- ``is_monophyletic``
- 检验给定的所有目标是否组成一个完成的子进化枝——即,存在一个进化枝满足:它的
末端节点和给定的目标是相同的集合。目标需要时树中的末端节点。为方便起见,若
给定目标是一个单系(monophyletic),这个方法将返回它们的共同祖先(MCRA)(
而不是
True),否则将返回False。 - ``is_parent_of``
- 若目标是这个树的后代(descendant)则为True——不必为直接后代。检验一个进化枝的
直接后代,只需要用简单的列表成员检测方法:
if subclade in clade: ... - ``is_preterminal``
- 若所有的直接后代都为末端则为True;否则任何一个直接后代不为末端则为False。
13.4.3 修改类方法¶
这些方法都在原地对树进行修改,所以如果你想保持原来的树不变,你首先要使用Python的
copy 模块对树进行完整的拷贝:
tree = Phylo.read('example.xml', 'phyloxml')
import copy
newtree = copy.deepcopy(tree)
- ``collapse``
- 从树中删除目标,重新连接它的子代(children)到它的父亲节点(parent)。
- ``collapse_all``
- 删除这个树的所有后代(descendants),只保留末端节点(terminals)。 分支长度被保留,即到每个末端节点的距离保持不变。如指定一个目标(见上), 只坍塌(collapses)和指定匹配的内部节点。
- ``ladderize``
- 根据末端节点的个数,在原地对进化枝(clades)进行排序。越深的进化枝默认被放到最后,
使用
reverse=True将其放到最前。 - ``prune``
- 从树中修剪末端进化枝(terminal clade)。如果分类名(taxon)来自一个二叉枝(bifurcation), 连接的节点将被坍塌,它的分支长度将被加到剩下的末端节点上。这可能不再是一个有意义的值。
- ``root_with_outgroup``
使用包含给定目标的外群进化枝(outgroup clade)重新确定树的根节点,即外群的共同祖先。该方法 只在Tree对象中能用,不能用于Clade对象。
如果外群和self.root一致,将不发生改变。如果外群进化枝是末端(即一个末端节点被作为外群),一个 新的二叉根进化枝将被创建,且到给定外群的分支长度为0。否则,外群根部的内部节点变为整个树的一个 三叉根。如果原先的根是一个二叉,它将被从树中遗弃。
在所有的情况下,树的分支长度总和保持不变。
- ``root_at_midpoint``
- 重新选择树中两个最远的节点的中点作为树的根。(这实际上是使用
root_with_outgroup函数。) - ``split``
- 产生 n (默认为2)个 新的后代。在一个物种树中,这是一个物种形成事件。新的进化枝拥有给定的
branch_length以及和这个进化枝的根相同的名字,名字后面包含一个整数后缀(从0开始计数)—— 例如,分割名为“A”的进化枝将生成子进化枝“A0”和“A1”。
查看Biopython维基的Phylo页面 (http://biopython.org/wiki/Phylo) 以获得更多已有方法的使用示例。
13.4.4 PhyloXML树的特性¶
phyloXML文件格式包含用来注释树的,采用额外数据格式和图像提示的字段。
参加Biopython维基上的PhyloXML页面 (http://biopython.org/wiki/PhyloXML) 以查看关于使用PhyloXML提供的额外注释特性的描述和例子。
13.5 运行外部程序¶
尽管Bio.Phylo本身不从序列比对推断进化树,但这里有一些第三方的程序可以使用。
他们通过 Bio.Phylo.Applications 模块获得支持,使用和 Bio.Emboss.Applications 、
Bio.Align.Applications 以及其他模块相同的通用框架。
Biopython 1.58引入了一个PhyML的打包程序(wrapper)
(http://www.atgc-montpellier.fr/phyml/)。
该程序接受一个 phylip-relaxed 格式(它是Phylip格式,然而没有对分类名称的10个字符的限制)
的比对输入和多种参数。一个快速的例子是:
>>> from Bio import Phylo
>>> from Bio.Phylo.Applications import PhymlCommandline
>>> cmd = PhymlCommandline(input='Tests/Phylip/random.phy')
>>> out_log, err_log = cmd()
这生成一个树文件盒一个统计文件,名称为:
[input filename]_phyml_tree.txt 和
[input filename]_phyml_stats.txt. 树文件的格式是Newick格式:
>>> tree = Phylo.read('Tests/Phylip/random.phy_phyml_tree.txt', 'newick')
>>> Phylo.draw_ascii(tree)
一个类似的RAxML打包程序 (http://sco.h-its.org/exelixis/software.html) 也已经被添加到Biopython 1.60中。
注意,如果你系统中已经安装了EMBOSS的Phylip扩展,一些常用的Phylip程序,包括 dnaml 和 protml
已经通过 Bio.Emboss.Applications 中的EMBOSS打包程序被支持。参见章节 6.4
以查看使用这些程序的例子和提示。
13.6 PAML整合¶
Biopython 1.58引入了对PAML的支持 (http://abacus.gene.ucl.ac.uk/software/paml.html), 它是一个采用最大似然法(maximum likelihood)进行系统进化分析的程序包。目前,对程序codeml、baseml和yn00的支持 已经实现。由于PAML使用控制文件而不是命令行参数来控制运行时选项,这个打包程序(wrapper)的使用格式和Biopython 的其他应用打包程序有些差异。
一个典型的流程是:初始化一个PAML对象,指定一个比对文件,一个树文件,一个输出文件和工作路径。下一步,运行时
选项通过 set_options() 方法或者读入一个已有的控制文件来设定。最后,程序通过 run() 方法来运行,输出文件
将自动被解析到一个结果目录。
下面是一个codeml典型用法的例子:
>>> from Bio.Phylo.PAML import codeml
>>> cml = codeml.Codeml()
>>> cml.alignment = "Tests/PAML/alignment.phylip"
>>> cml.tree = "Tests/PAML/species.tree"
>>> cml.out_file = "results.out"
>>> cml.working_dir = "./scratch"
>>> cml.set_options(seqtype=1,
... verbose=0,
... noisy=0,
... RateAncestor=0,
... model=0,
... NSsites=[0, 1, 2],
... CodonFreq=2,
... cleandata=1,
... fix_alpha=1,
... kappa=4.54006)
>>> results = cml.run()
>>> ns_sites = results.get("NSsites")
>>> m0 = ns_sites.get(0)
>>> m0_params = m0.get("parameters")
>>> print m0_params.get("omega")
已有的输出文件也可以通过模块的 read() 方法来解析:
>>> results = codeml.read("Tests/PAML/Results/codeml/codeml_NSsites_all.out")
>>> print results.get("lnL max")
这个新模块的详细介绍目前在Biopython维基上可以看到: http://biopython.org/wiki/PAML
13.7 未来计划¶
Bio.Phylo 目前还在开发中,下面是我们可能会在将来的发布版本中添加的特性:
- 新方法
- 通常用来操作Tree和Clade对象的有用方法会首先出现在Biopython维基上,这样常规用户 就能在我们添加到Bio.Phylo之前测试这些方法,看看它们是否有用: http://biopython.org/wiki/Phylo_cookbook
- Bio.Nexus port
这个模块的大部分是在2009年NESCent主办的谷歌编程夏令营中写的,作为实现Python对phyloXML数据格式(见 13.4.4 )支持的一个项目。对Newick和Nexus格式的支持,已经通过导入Bio.Nexus模块 的一部分被添加到Bio.Phylo使用的新类中。
目前,Bio.Nexus包含一些还没有导入到Bio.Phylo类中的有用的特性——特别是,计算一致树(consensus tree)。 如果你发现某些功能Bio.Phylo中没有,试试在Bio.Nexus中能不能找到。
我们乐意接受任何增强该模块功能和使用性的建议;如果有,只需要通过邮件列表或我们的bug数据库让我们知道。